






IMPORTANT SAFETY INFORMATION
Warning:
WARNING: INFUSION REACTIONS AND CARDIOPULMONARY ARREST
Infusion Reactions - ERBITUX can cause serious and fatal infusion reactions. Severe (Grades 3 and 4) infusion reactions occurred in 2.2% of patients receiving ERBITUX in clinical trials.
- The risk of anaphylactic reactions may be increased in patients with a history of tick bites, red meat allergy, or in the presence of IgE antibodies directed against galactose-α-1,3-galactose (alpha-gal). Consider testing patients for alpha-gal IgE antibodies using FDA-cleared methods prior to initiating ERBITUX. Negative results for alpha-gal antibodies do not rule out the risk of severe infusion reactions.
- Approximately 90% of the severe infusion reactions occurred with the first infusion of ERBITUX despite premedication with antihistamines.
- Serious infusion reactions, requiring immediate medical intervention, included symptoms of rapid onset of airway obstruction (bronchospasm, stridor, hoarseness), hypotension, shock, loss of consciousness, myocardial infarction, and/or cardiac arrest. Immediately interrupt and permanently discontinue ERBITUX infusion for serious infusion reactions.
- Caution must be exercised with every ERBITUX infusion as infusion reactions may occur during or several hours following completion of the infusion.
- Premedicate with a histamine-1 (H1) receptor antagonist as recommended.
- Monitor patients for at least 1 hour following each ERBITUX infusion in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis. In patients requiring treatment for infusion reactions, monitor for more than 1 hour to confirm resolution of the reaction. Interrupt the infusion and upon recovery, resume the infusion at a slower rate or permanently discontinue ERBITUX based on severity.
- Carefully consider the use of ERBITUX with radiation therapy, or with platinum-based therapy with fluorouracil, in head and neck cancer patients with a history of coronary artery disease, congestive heart failure, or arrhythmias.
- Closely monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX therapy.
Pulmonary Toxicity
- ERBITUX can cause interstitial lung disease (ILD). ILD, which was fatal in one case, occurred in <0.5% of 1570 patients receiving ERBITUX in clinical trials. Monitor patients for signs and symptoms of pulmonary toxicity. Interrupt or permanently discontinue ERBITUX for acute onset or worsening of pulmonary symptoms. Permanently discontinue ERBITUX for confirmed ILD.
Dermatologic Toxicities
-
ERBITUX can cause dermatologic toxicities, including acneiform rash, skin drying and fissuring, paronychial inflammation, infectious sequelae (e.g., S. aureus sepsis, abscess formation, cellulitis, blepharitis, conjunctivitis, keratitis/ulcerative keratitis with decreased visual acuity, cheilitis), and hypertrichosis
- Acneiform rash occurred in 82% of the 1373 patients who received ERBITUX across clinical trials. Severe (Grades 3 or 4) acneiform rash occurred in 10% of patients. Acneiform rash usually developed within the first 2 weeks of therapy; the rash lasted more than 28 days after stopping ERBITUX in most patients.
- Life-threatening and fatal bullous mucocutaneous disease with blisters, erosions, and skin sloughing has been observed in patients who received ERBITUX. It could not be determined whether these mucocutaneous adverse reactions were directly related to EGFR inhibition or to idiosyncratic immune-related effects (e.g., Stevens-Johnson syndrome or toxic epidermal necrolysis).
- Monitor patients receiving ERBITUX for dermatologic toxicities and infectious sequelae.
- Sun exposure may exacerbate these effects. Instruct patients to limit sun exposure during ERBITUX therapy.
- Withhold, reduce dose or permanently discontinue ERBITUX based on severity of acneiform rash or mucocutaneous disease.
Risks Associated with Use in Combination with Radiation and Cisplatin
- ERBITUX is not indicated for the treatment of SCCHN in combination with radiation and cisplatin.
- In a controlled study, 940 patients with locally advanced SCCHN were randomized 1:1 to receive either ERBITUX in combination with radiation therapy and cisplatin, or radiation therapy and cisplatin alone. The addition of ERBITUX resulted in an increase in the incidence of Grade 3 and 4 mucositis, radiation recall syndrome, acneiform rash, cardiac events, and electrolyte disturbances compared to radiation and cisplatin alone.
- Adverse reactions with fatal outcome were reported in 4% of patients in the ERBITUX combination arm and 3% in the control arm.
- In the ERBITUX arm, 2% experienced myocardial ischemia compared to 0.9% in the control arm.
- The addition of ERBITUX to radiation and cisplatin did not improve progression-free survival (the primary endpoint).
Hypomagnesemia and Accompanying Electrolyte Abnormalities
-
ERBITUX can cause hypomagnesemia. Hypomagnesemia occurred in 55% of 365 patients receiving ERBITUX in study CA225-025 and two other clinical trials in patients with colorectal cancer (CRC) or head and neck cancer, including Grades 3 and 4 in 6% to 17%. In EXTREME, where a cetuximab product was administered in combination with platinum-based therapy, the addition cetuximab to cisplatin and fluorouracil resulted in an increased incidence of hypomagnesemia of any grade (14%) and of Grade 3 or 4 hypomagnesemia (7%). Hypomagnesemia of any grade occurred in 4% of patients who received cetuximab, carboplatin, and fluorouracil. No patient experienced grade 3 or 4 hypomagnesemia. The onset of hypomagnesemia and accompanying electrolyte abnormalities can occur days to months after initiating ERBITUX.
- Monitor patients weekly during treatment for hypomagnesemia, hypocalcemia, and hypokalemia, and for at least 8 weeks following the completion of ERBITUX.
- Replete electrolytes as necessary.
Increased Tumor Progression, Increased Mortality, or Lack of Benefit in Patients with Ras-Mutant mCRC
- ERBITUX is not indicated for the treatment of patients with CRC that harbor somatic mutations in exon 2 (codons 12 and 13), exon 3 (codons 59 and 61), and exon 4 (codons 117 and 146) of either K-Ras or N-Ras and hereafter referred to as "Ras" or when the Ras status is unknown.
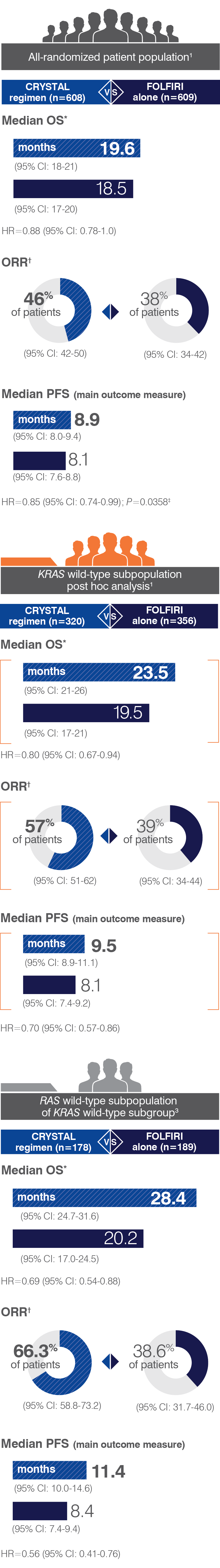
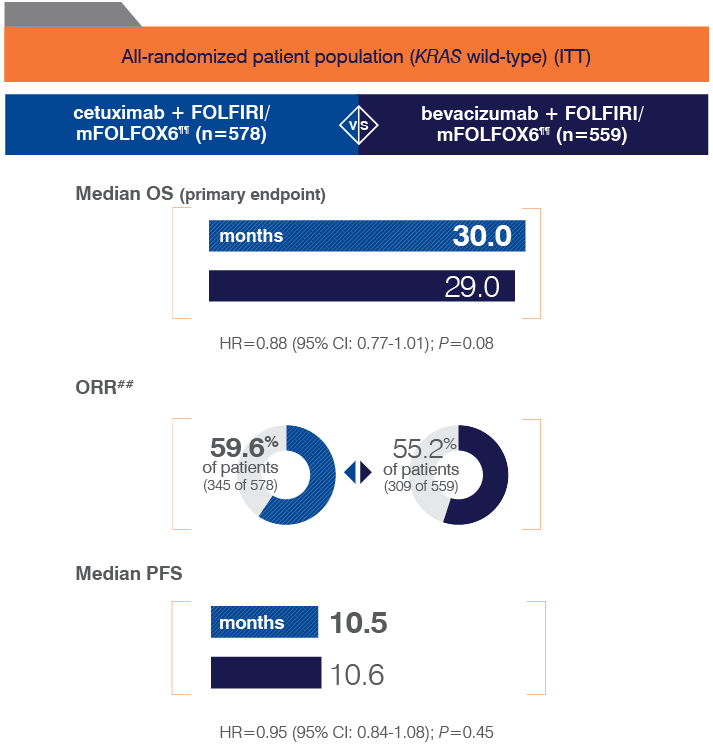
- Retrospective subset analyses of Ras-mutant and wild-type populations across several randomized clinical trials, including CRYSTAL, were conducted to investigate the role of Ras mutations on the clinical effects of anti-EGFR-directed monoclonal antibodies. Use of cetuximab in patients with Ras mutations resulted in no clinical benefit with treatment related toxicity. Confirm Ras mutation status in tumor specimens prior to initiating ERBITUX.
Embryo-Fetal Toxicity
- Based on animal data and its mechanism of action, ERBITUX can cause fetal harm when administered to a pregnant woman. There are no available data for ERBITUX exposure in pregnant women. In an animal reproduction study, intravenous administration of cetuximab once weekly to pregnant cynomolgus monkeys during the period of organogenesis resulted in an increased incidence of embryolethality and abortion. Disruption or depletion of EGFR in animal models results in impairment of embryo-fetal development including effects on placental, lung, cardiac, skin, and neural development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ERBITUX and for 2 months after the last dose of ERBITUX. Verify pregnancy status in females of reproductive potential prior to initiating ERBITUX.
Adverse Reactions
- The most common adverse reactions (all grades; incidence ≥25%) seen in patients with carcinomas of the head and neck receiving ERBITUX in combination with radiation therapy (n=208) versus radiation alone (n=212) (BONNER) were acneiform rash (87% vs 10%), radiation dermatitis (86% vs 90%), weight loss (84% vs 72%), asthenia (56% vs 49%), nausea (49% vs 37%), increased alanine transaminase (43% vs 21%), increased aspartate transaminase (38% vs 24%), increased alkaline phosphatase (33% vs 24%), fever (29% vs 13%), emesis (29% vs 23%), pharyngitis (26% vs 19%) and dehydration (25% vs 19%). The most common grade 3 and 4 adverse reactions for ERBITUX in combination with radiation therapy (≥10%) versus radiation alone included: radiation dermatitis (23% vs 18%), acneiform rash (17% vs 1%), and weight loss (11% vs 7%). The overall incidence of late radiation toxicities (any grade) was higher for patients receiving ERBITUX in combination with radiation therapy, versus radiation therapy alone. The following sites were affected: salivary glands (65% vs 56%), larynx (52% vs 36%), subcutaneous tissue (49% vs 45%), mucous membrane (48% vs 39%), esophagus (44% vs 35%), skin (42% vs 33%). The incidence of Grade 3 or 4 late radiation toxicities was similar between radiation therapy alone and the ERBITUX with radiation treatment groups.
- The most common adverse reactions (all grades; incidence ≥25%) seen in patients with carcinomas of the head and neck receiving a cetuximab product in combination with platinum-based therapy and fluorouracil (CT) (n=219) versus CT alone (n=215) (EXTREME) were acneiform rash (70% vs 2%), nausea (54% vs 47%), infection (44% vs 27%), rash (28% vs 2%), diarrhea (26% vs 16%) and anorexia (25% vs 14%). The most common grade 3 and 4 adverse reaction for a cetuximab product in combination with CT (≥10%) versus CT alone was infection (11% vs 8%). Because ERBITUX provides approximately 22% higher exposure relative to the cetuximab product used in EXTREME, the data provided above may underestimate the incidence and severity of adverse reactions anticipated with ERBITUX for this indication. However, the tolerability of the recommended dose is supported by safety data from additional studies of ERBITUX.
- The most common adverse reactions (all grades; incidence ≥25%) seen in patients with K-Ras wild-type, EGFR-expressing mCRC treated with a cetuximab product in combination with FOLFIRI (n=317) versus FOLFIRI alone (n=350) (CRYSTAL) were acne-like rash (86% vs 13%), diarrhea (66% vs 60%), neutropenia (49% vs 42%), rash (44% vs 4%), stomatitis (31% vs 19%), anorexia (30% vs 23%), dermatitis acneiform (26% vs <1%) and pyrexia (26% vs 14%). The most common grade 3 and 4 adverse reactions (≥10%) included: neutropenia (31% vs 24%), acne-like rash (18% vs <1%), and diarrhea (16% vs 10%). ERBITUX provides approximately 22% higher exposure compared to the cetuximab product used in CRYSTAL; however, the safety data from CRYSTAL is consistent in incidence and severity of adverse reactions with those seen for ERBITUX in this indication.
- The most common adverse reactions (all grades; incidence ≥25%) seen in patients with K-Ras wild-type, EGFR-expressing mCRC treated with ERBITUX + best supportive care (BSC) (n=118) versus BSC alone (n=124) (CA225-025) were rash/desquamation (95% vs 21%), fatigue (91% vs 79%), nausea (64% vs 50%), pain-other (59% vs 37%), dry skin (57% vs 15%), constipation (53% vs 38%), dyspnea (49% vs 44%), pruritus (47% vs 11%), neuropathy-sensory (45% vs 38%), diarrhea (42% vs 23%), vomiting (40% vs 26%), headache (38% vs 11%), infection without neutropenia (38% vs 19%), other-dermatology (35% vs 7%), stomatitis (32% vs 10%), nail changes (31% vs 4%), cough (30% vs 19%), insomnia (27% vs 13%) and fever (25% vs 16%). The most common grade 3 and 4 adverse reactions (≥10%) included: fatigue (31% vs 29%), pain-other (18% vs 10%), rash/desquamation (16% vs 1%), dyspnea (16% vs 13%), other-gastrointestinal (12% vs 5%), and infection without neutropenia (11% vs 5%).
- The most common adverse reactions (all grades) seen in patients with EGFR-expressing recurrent mCRC (n=354) treated with ERBITUX plus irinotecan in clinical trials (CP02-9923 and BOND) were acneiform rash (88%), asthenia/malaise (73%), diarrhea (72%), and nausea (55%). The most common grade 3-4 adverse reactions included: diarrhea (22%), leukopenia (17%), asthenia/malaise (16%), and acneiform rash (14%).
- The most common adverse reactions (all grades; incidence ≥25%) seen in patients with BRAF V600E mutation-positive mCRC treated with ERBITUX in combination with encorafenib (N=216) versus ERBITUX with irinotecan or ERBITUX with FOLFIRI (N=193) (BEACON) were fatigue (51% vs 50%), nausea (34% vs 41%), diarrhea (33% vs 48%), dermatitis acneiform (32% vs 43%), abdominal pain (30% vs 32%), decreased appetite (27% vs 27%), arthralgia (27% vs 3%) and rash (26% vs 26%). Other clinically important adverse reactions occurring in <10% of patients who received ERBITUX in combination with encorafenib were pancreatitis. The most common laboratory abnormalities (all grades; incidence ≥20%) seen in patients receiving ERBITUX in combination with encorafenib versus ERBITUX with irinotecan or ERBITUX with FOLFIRI (BEACON) were anemia (34% vs 48%) and lymphopenia (24% vs 35%).
Use in Specific Populations
- Lactation: There is no information regarding the presence of ERBITUX in human milk, the effects on the breastfed infant, or the effects on milk production. Human IgG antibodies can be excreted in human milk. Because of the potential for serious adverse reactions in nursing infants from ERBITUX, advise women not to breastfeed during treatment with ERBITUX and for at least 2 months after the last dose of ERBITUX.
- Pediatric Use: The safety and effectiveness of ERBITUX in pediatric patients have not been established. No new safety signals were identified in pediatric patients when ERBITUX in combination with irinotecan was administered in an open-label, single-arm dose-finding study in 27 patients with refractory solid tumors aged 1 to 12 years old and in 19 patients aged 13 to 18 years old.
- Geriatric Use: In SCCHN clinical studies of ERBITUX there were insufficient number of patients >65 years of age to determine whether they respond differently from younger patients.
CE HCP ISI_ALL 14SEP2022